Home » Equipos Médicos » Certificaciones y precauciones para la exportación de dispositivos médicos a Malasia.
Equipos MédicosExportar a Malasia requiere cumplir con ciertas certificaciones y requisitos específicos. A continuación se presentan las certificaciones y consideraciones necesarias para exportar dispositivos médicos a Malasia:
I. Legislación y autoridad competente en dispositivos médicos
La ley de gestión de dispositivos médicos en Malasia es la Ley de Gestión de Dispositivos Médicos (2012), cuyo marco básico se asemeja bastante al de las regulaciones estadounidenses para dispositivos médicos. Los organismos encargados de la gestión de dispositivos médicos incluyen la Autoridad de Dispositivos Médicos (Medical Device Authority, MDA) del Ministerio de Salud y la Agencia de Licencias de Energía Atómica del Ministerio de Ciencia y Tecnología. La Autoridad de Dispositivos Médicos del Ministerio de Salud se encarga de gestionar todos los dispositivos médicos, excepto los dispositivos médicos radiactivos y los dispositivos médicos de segunda mano, mientras que la Agencia de Licencias de Energía Atómica se encarga de gestionar los dispositivos médicos radiactivos y los dispositivos médicos de segunda mano.
II. Proceso de acceso al mercado para dispositivos médicos importados de Malasia
(1) Definición y clasificación de los dispositivos médicos en Malasia
Los dispositivos médicos se definen como cualquier instrumento, aparato, implemento, máquina, implante, reactivo o calibrador in vitro, software, material, etc., utilizado solo o en combinación, que tenga los siguientes fines. Pueden utilizarse para diagnosticar, prevenir, vigilar, tratar o aliviar enfermedades y lesiones, investigar procesos anatómicos o fisiológicos, apoyar o mantener la vida, desinfectar dispositivos y proporcionar información para fines médicos o de diagnóstico mediante el examen in vitro de muestras obtenidas del cuerpo humano. Sin embargo, si cumplen estas funciones mediante procesos farmacológicos, inmunológicos u otros similares, no pueden definirse como dispositivos médicos. Según la normativa de gestión de dispositivos médicos de Malasia, estos se clasifican en cuatro categorías según el riesgo, de menor a mayor: clase A, clase B, clase C y clase D. Entre ellas, los dispositivos médicos de clase A presentan el riesgo más bajo, las clases B y C un riesgo intermedio y la clase D el riesgo más alto.
Producto de categoría A: bajo riesgo, como el contador de células.
Producto de la categoría B: riesgo bajo a moderado, como el analizador de orina.
Categoría C: riesgo medio-alto, como los electrocardiógrafos.
Productos de la categoría D: alto riesgo, como las máquinas de hemodiálisis.
(2) Clasificación y registro de reactivos para diagnóstico in vitro
Los reactivos para diagnóstico in vitro (in-vitro diagnostic, IVD) son dispositivos que se utilizan solos o en combinación única o principalmente para proporcionar información diagnóstica o de monitoreo para el cuerpo humano, incluyendo reactivos, calibradores, recipientes para muestras, entre otros. Según la Ley de Gestión de Dispositivos Médicos, los productos IVD se clasifican en cuatro categorías: A, B, C y D, según el nivel de riesgo que representan para la salud individual y pública. A continuación, se presentan los niveles de riesgo y ejemplos de productos para cada categoría:

Para el registro de IVD, es necesario seguir los criterios aplicables específicos establecidos en la Ley de Gestión de Dispositivos Médicos y llevar a cabo el registro siguiendo los pasos correspondientes.

III. Proceso de registro de dispositivos médicos importados de Malasia
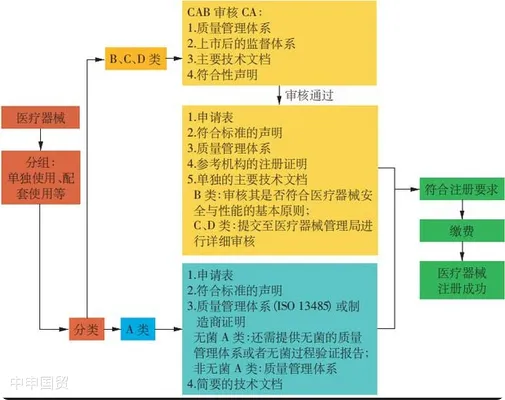
(1) Productos de la categoría A
Para los productos de la categoría A, el representante autorizado local (Authorized Representative, AR) puede solicitar el registro ante la Administración de Dispositivos Médicos del Ministerio de Salud sin necesidad de la aprobación de un Organismo de Evaluación de la Conformidad (Conformity Assessment Body, CAB) autorizado por la MDA. El solicitante debe presentar documentos como el certificado ISO 13485 del fabricante, los informes de prueba y las etiquetas.
(2) Productos de las categorías B, C y D
Para los productos de las categorías B, C y D, el representante autorizado local de dispositivos médicos debe presentar un informe técnico y someter los documentos técnicos a revisión por parte de una entidad de evaluación de la conformidad. Los dispositivos médicos ya aprobados y comercializados en países de referencia (como Australia, Canadá, la Unión Europea, Japón y Estados Unidos) pueden ser evaluados mediante un procedimiento simplificado. Durante la evaluación, se deben entregar a la entidad de evaluación de la conformidad documentos como el certificado ISO y el certificado CE. Una vez aprobada la evaluación, la entidad de evaluación de la conformidad emitirá el correspondiente certificado. El expediente final de registro del dispositivo, que incluye la Plantilla Común de Expediente de Solicitud (Common Submission Dossier Template, CSDT), el certificado de la entidad de evaluación de la conformidad y la documentación de solicitud, se presentará en línea de forma electrónica ante la Administración de Dispositivos Médicos del Ministerio de Salud para su revisión y aprobación final. Tanto el certificado de la entidad de evaluación de la conformidad como el certificado de registro del dispositivo deberán renovarse cada 5 a?os.
IV. Precauciones
Durante el proceso de exportación de dispositivos médicos a Malasia, se deben tener en cuenta los siguientes aspectos:
(1) Actualmente, ya no se requiere el Certificado de Libre Venta (Free Sales Certificate, FSC) para el registro en Malasia.
(2) La solicitud de registro debe presentarse ante la oficina de representación local designada en Malasia.
(3) Cada producto solo puede tener un titular de licencia de producto (es decir, el titular de la licencia).
(4) La licencia del producto puede transferirse a otro titular.
(5) Todos los fabricantes deberán obtener la certificación ISO 13485 como requisito indispensable para solicitar el registro.
En resumen, la exportación de dispositivos médicos a Malasia debe cumplir con los requisitos de registro establecidos en la Ley de Gestión de Dispositivos Médicos. El proceso específico incluye la determinación de la clasificación, la revisión y presentación de la documentación técnica, la certificación por parte de un organismo de evaluación de la conformidad y la aprobación final del registro en línea. Antes de solicitar el registro, se debe conocer detalladamente la normativa y los requisitos pertinentes, y asegurarse de cumplir con los estándares de exportación de dispositivos médicos de Malasia.
Recomendaciones relacionadas